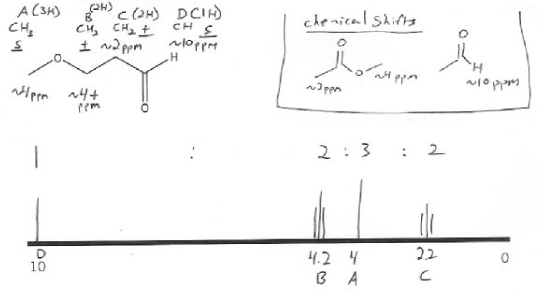
There are four types of protons (hydrogens) in the compound below, so we would expect to see four peaks (A, B, C, and D).
A is not adjacent to any other types of protons, so its multiplicity is 1, also called singlet (s). ( n=0, so n + 1 = 0 + 1 = 1). A also is a CH3, so it will have an integration of 3. Finally, A's proton are on a carbon adjacent to an oxygen, so its chemical shit will be about ~4 ppm.
B is a CH2 so its integration is 2. It's adjacent to a 2 hydrogens (a CH2 ) so its multiplicty is 3, also called a triplet (t). (n=2, so n + 1 = 3). B is also adjacent to oxygen, so it will be close to ~4 ppm, but a little further downfield than A because it's more substituted than A.
C is also a CH2 so its integration is 2. It's also adjacent to 2 hydrogens so it's a triplet (n+1 rule). C's hydrogens are on a carbon adjacent to a carbonyl (C=O), so its chemical shift will be around ~2 ppm.
D is an aldehyde proton, which is always a singlet with near ~10 ppm. It's only one proton, so its integration is 1.
Note: notice that the aldehyde proton doesn't couple to another other protons! So even through C is surrounded by a CH2 on its left and an aldehyde proton on its right, when determining multiplicity we only count the CH2, and not the aldehyde proton.